Download

Hyperbranched polymers are macromolecules with irregular structure, they have polydispersity of parameters such as molecular weight and degree of branching. Especially when the hyperbranched polymer is grafted with a linear arm to form a hyperbranched multi-arm copolymer, in addition to the polydispersity of the hyperbranched core, the grafted arms also have molecular weight distribution and graft ratio. However, at present, the modeling of hyperbranched polymers is mainly depended on commercial software, which usually treats hyperbranched polymers as dendrimers, and does not consider polydispersity at all. Obviously, such a model is inconsistent with the actual situation. To solve this problem, our group developed a new hyperbranched polymer modeling software HBP Builder. The software combines the characteristics of coarse-grained and all-atom simulations, comprehensively considering the polydispersity of hyperbranched polymers and hyperbranched multi-arm copolymers, including molecular weight distribution, degree of branching, grafting rate, etc., which can truly describe the hyperbranched polymers synthesized in the experiment. And the work efficiency of the software is extremely high, as long as the average molecular weight, molecular weight distribution coefficient, average degree of branching and degree of branching distribution coefficient of the polymer are input (these data can be measured through experiments), it can generate thousands of coarse-grained models of hyperbranched polymers that meet the requirements within a few seconds. More importantly, the software can quickly transform the obtained coarse-grained model into an all-atom model, and further connect with the experimental data. In addition, HBP Builder software can also be used for dendrimer and linear polymer modeling with good scalability. The development of the software cleared the obstacles for the theoretical simulation of hyperbranched polymers. This work has been published in Scientific reports (Sci. Rep. 2016, 6, 26264).【click to download】
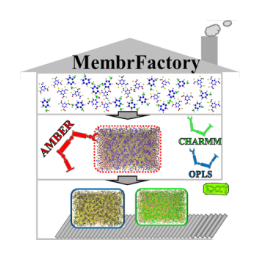
Molecular dynamics simulation can help people understand the physical and chemical processes that occur in polyamide reverse osmosis membranes from a molecular perspective, and reveal the transmission and salt rejection mechanisms during reverse osmosis process. At present, the methods that can be used to construct an all-atom polyamide reverse osmosis membrane model are very limited and are mainly divided into two categories: (i) starting from a linear polyamide chain, and constructing the model through random cross-linking reactions between chains and (ii) With the monomer as the initial state, the model is constructed through the continuous reaction between the monomers. The latter method has gained more recognition because it is closer to the actual film formation process. In recent years, in the field of research on polyamide reverse osmosis composite membranes, many valuable results have been obtained through molecular dynamics simulations. However, due to the poor versatility of current modeling methods, the types of reverse osmosis membranes studied are relatively simple, mainly focusing on the research of FT-30 membranes. With the emergence of more and more new polyamide reverse osmosis membrane materials, how to quickly and effectively construct polyamide reverse osmosis membrane models with different structures is crucial for the application of molecular dynamics simulation in the field of polyamide reverse osmosis membrane. In order to solve this problem, our group has developed a universal polyamide reverse osmosis membrane modeling tool "MembrFactory", which is compatible with different force fields and can be freely selected the structure of the building monomers. On this basis, users can further study the relationship between the structure and performance of membranes constructed by different monomers through molecular dynamics simulations, thereby providing theoretical guidance for the subsequent modification of the membrane structure. This work has been published in the Journal of Computational Chemistry (J. Comput. Chem. 2019, 40, 2432-2438).【click to download】

沪交ICP备20190229 版权所有© 上海交通大学
地址:上海市东川路800号化学化工学院 联系电话:86-21-54742665